


Every year millions of Americans (no one knows exactly how many) volunteer to be human subjects in medical research that compares a new treatment with an old one—or when there is no existing treatment, with a placebo. By something like a coin toss, some volunteers are assigned to get the new treatment (the experimental group), while others get the old one (the control group). This type of research is termed a clinical trial, and at any given time there are thousands underway in the US. Most are sponsored by makers of prescription drugs or medical devices, but many are sponsored by the government, mainly the National Institutes of Health (NIH). A growing number are conducted offshore, particularly in countries with autocratic governments, where they are easier and cheaper to do.
The first modern clinical trial was published only sixty-seven years ago, in 1948. Sponsored by the British Medical Research Council, the trial compared streptomycin, a new antibiotic, with bedrest alone in patients with tuberculosis. (Streptomycin proved better, and became part of the usual treatment for this disease.) Before that, human experimentation was fairly haphazard; subjects were treated in one way or another to see how they fared, but there was usually no comparison group. Even when there was, the comparison lacked the rigorous methods of modern clinical trials, which include randomization to make sure the two groups are similar in every way except the treatment under study. After the streptomycin study, carefully designed clinical trials soon became the scientific standard for studying nearly any new medical intervention in human subjects.1
Patients with serious medical conditions are often eager to enroll in clinical trials in the mistaken belief that experimental treatments are likely to be better than standard treatments (most turn out to be no better, and often worse).2 Healthy volunteers, on the other hand, are motivated by some combination of the modest payments they receive and the altruistic desire to contribute to medical knowledge.
Given the American faith in medical advances (the NIH is largely exempt from the current disillusionment with government), it is easy to forget that clinical trials can be risky business. They raise formidable ethical problems since researchers are responsible both for protecting human subjects and for advancing the interests of science. It would be good if those dual responsibilities coincided, but often they don’t. On the contrary, there is an inherent tension between the search for scientific answers and concern for the rights and welfare of human subjects.
Consider a hypothetical example. Suppose researchers want to test a possible vaccine against HIV infection. Scientifically, the best way to do that would be to choose healthy participants for a trial, give the vaccine to half of them, and then inject all of them with HIV and compare the infection rates in the two groups. If there were significantly fewer HIV cases in the vaccinated group than in the unvaccinated group, that would prove that the vaccine worked. Such a trial would be simple, fast, and conclusive. In short order, we would have a clear answer to the question of whether the vaccine was effective—an answer that might have enormous public health importance and save many lives.
Yet everyone today would agree that such a trial would be unethical. If asked why, most people would probably say that people should not be treated like guinea pigs—that is, they should not be used merely as a means to an end. (And, of course, no fully informed person would volunteer for such a trial.) There is an instinctive revulsion against deliberately infecting human subjects with a life-threatening disease, no matter how important the scientific question.
So in practice, the researchers in this hypothetical study would have to make scientific concessions for ethical reasons. Since they would be prohibited from injecting HIV, they would simply have to wait to see how many in each group (vaccinated and unvaccinated) got infected in the usual course of their lives. That could take many years and a very large number of subjects. Even if the researchers chose subjects at high risk of becoming infected with HIV—say, intravenous drug users—many people would have to be followed for a long time to accumulate the number of infections needed to permit a statistically valid comparison between the vaccinated and unvaccinated groups. Even then, the results might be hard to interpret, because of differences in exposures between the two groups. In short, doing an ethical trial would be far less efficient and conclusive—and much more expensive—than simply injecting the virus.
Conducting the trial ethically and more slowly might not just mean a loss of scientific efficiency. If the vaccine turned out to be effective, it could also mean a loss of lives—the lives of all those throughout the world who contracted HIV infection for want of a vaccine during the extra time it took to do an ethical trial. There would have been a trade-off between the welfare of participants in the trial and the welfare of the far larger number of people who would benefit from finding an effective vaccine quickly. Either human subjects would suffer by being deliberately exposed to HIV infection in an unethical trial, or future patients would suffer by having been deprived of a vaccine while an ethical trial was ongoing.
Advertisement
This hypothetical example of the tension between science and society, on the one hand, and ethics, on the other, is admittedly extreme. Nearly everyone would agree on the right course of action in this case; they would reject the utilitarian claim that injecting human subjects with HIV would do the greatest good for the greatest number. Yet there have been many real experiments over the years involving no less extreme choices in which researchers sacrificed the welfare of human subjects to the interests of science and future patients and believed they were right to do so.
The most horrifying and grotesque of these were the medical experiments carried out by Nazi Germany during World War II on inmates in concentration camps.3 Although it is difficult to believe now, the people who designed these experiments—among them some of the most prominent physicians in Germany at the time—did have a purpose. They wanted to gain information that could save the lives of German troops in battle. In one experiment in Dachau, for example, their aim was to find the maximum altitude at which it would be safe for pilots to parachute from stricken planes. To that end, they put inmates in vacuum chambers that could duplicate progressively lower air pressure, up to the equivalent of an altitude of about 68,000 feet. About 40 percent of the victims died from lack of oxygen during these hideous experiments.
In another experiment, the researchers wanted to study how long pilots who had parachuted into the frigid North Sea could survive. They immersed victims in tanks of ice water for up to three hours, and many froze to death. The Nazis also tested a typhus vaccine by administering it to some, but not all, of a group of prisoners. They then infected them all with typhus to compare the death rate—an experiment identical in design to my hypothetical example.
At the end of the war, a series of war crimes trials were held by the victorious Allies in Nuremberg, Germany. One of them, presided over by American judges in a US military tribunal, was known as the Doctors’ Trial. In that trial, which began in December 1946, the twenty-three surviving researchers (twenty of them physicians) responsible for the medical experiments in the concentration camps were accused of war crimes and crimes against humanity. In their defense, they presented a litany of excuses. Two of them are particularly important, because in modified form they have to this day been used repeatedly in defense of unethical research.
First, the Nazi doctors offered the utilitarian argument that their research would save more lives than it cost. They went on to say that this justification was all the more convincing, because the lives at stake were troops fighting for the very survival of their country. Extreme circumstances demand extreme action, they said. Second, the Nazi doctors pointed out that many of their human subjects were condemned to death anyway (for “crimes” such as being a Gypsy or a Jew). Those selected for medical experimentation might even have lived longer than they otherwise would.
The Nazi Doctors’ Trial resulted in the conviction in August 1947 of sixteen of the twenty-three defendants, seven of whom were hanged and nine imprisoned. As part of the judgment, the court issued the celebrated Nuremberg Code in 1947—the first, shortest, and in many ways most uncompromising of the major ethical codes and regulations for the conduct of medical research on humans. Although the code had no legal authority in any country, it had great influence on ideas about human experimentation, and subsequent international codes and legislation.
The first provision of the Nuremberg Code is unqualified: “The voluntary consent of the human subject is absolutely essential.” No exceptions are permitted. Given the Nazi experience, the reason for this is obvious. Experimentation on children and others not capable of deciding for themselves is prohibited, since the code requires volunteers to have the “legal capacity” to consent. Furthermore, the code is clear that consent must be given by subjects who are fully informed. It states:
Before the acceptance of an affirmative decision by the experimental subject, there should be made known to him the nature, duration, and purpose of the experiment; the method and means by which it is to be conducted; all inconveniences and hazards reasonably to be expected; and the effects upon his health or person, which may possibly come from his participation in the experiment.
Other provisions are less absolute, and dependent on the judgment of researchers. One is the requirement that experiments should be “such as to yield fruitful results for the good of society” and “not random and unnecessary in nature”—that is, research should not be undertaken in the first place if it is not important. (Even with the most generous interpretation, that provision is now routinely violated, particularly by companies that sponsor research primarily to increase their sales.)
Advertisement
The code also stipulates that “the degree of risk to be taken should never exceed that determined by the humanitarian importance of the problem to be solved by the experiment,” and that researchers should terminate experiments if they believe continuing them would be “likely to result in injury, disability, or death to the experimental subject[s].” According to the Nuremberg Code, then, informed consent is necessary, but not sufficient.
Was the issuance of the Nuremberg Code the end of unethical medical research? Not at all. In fact, over about thirty years, from 1944 to 1974, the US government conducted multiple experiments in which people were deliberately exposed to radiation without their knowledge or consent. The aim was to study the effects of testing nuclear weapons. To cite only two examples, the testicles of state prisoners in Oregon and Washington were irradiated to study the effects on sperm production, and terminally ill patients in a Cincinnati hospital underwent irradiation of their entire bodies to learn about its dangers to military personnel.4 Note the irony that these experiments were taking place during the Nuremberg trials, and involved a similar justification—namely, that extreme circumstances, in this case the cold war, demand extreme actions.
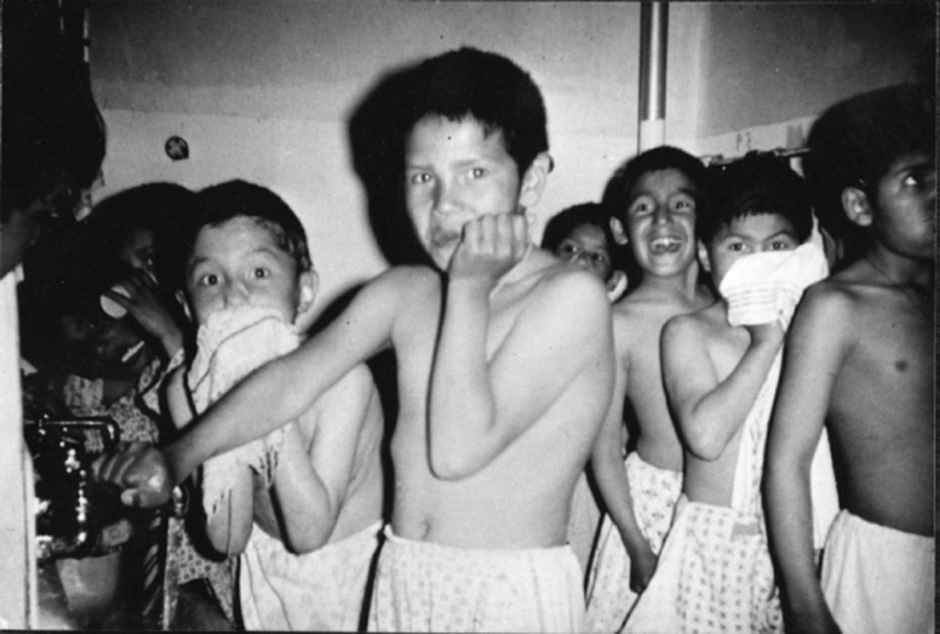
From 1956 to 1972, a study was conducted at the Willowbrook State School for the Retarded in New York, in which the children there were deliberately infected with hepatitis to study the natural course of the disease and its treatment. The justification was that the sanitation in the facility was so poor that nearly all of these children would have contracted the illness anyway.5 Note the similarity to the Nazi doctors’ excuse that many of the inmates in the concentration camps were condemned to death anyway.
Still, with the promulgation of the Nuremberg Code, there were now some broadly accepted principles, despite such egregious violations. Nevertheless, for all its strengths, it was felt that the Nuremberg Code needed change, mainly because the absolute requirement for informed consent by competent adults was widely seen as too stringent. There was also a need to address some of the special issues raised by the design of the new clinical trials introduced in the 1940s. How should risks and benefits to the experimental and control groups be balanced?
In 1964, the World Medical Association (WMA), which consists of a group of national medical societies, including the American Medical Association (AMA), issued the first Declaration of Helsinki. It has undergone seven revisions since then, most recently in 2013. Like the Nuremberg Code, the Declaration of Helsinki has no legal authority, but it rapidly became the new ethical standard by which clinical trials were judged, and for many years, the NIH and FDA explicitly required that research they oversaw conformed with its principles. From the first, the Declaration of Helsinki weakened the Nuremberg Code’s absolute requirement for informed consent by requiring consent only “if at all possible,” and permitting research on children and others not capable of deciding for themselves, if parents or other legal proxies consented. Nevertheless, the first revision (1975) contained this unequivocal statement, “Concern for the interests of the subject must always prevail over the interest of science and society,” and it also recommended oversight by independent committees.
But the Declaration of Helsinki foundered with the fifth revision in 2000 and its aftermath. The problem arose in the wake of heated debate about some clinical trials sponsored by the NIH and the Centers for Disease Control and Prevention (CDC) in developing countries.
First some background: in a typical clinical trial, a new treatment is compared with an old one. In order to give “precedence to the well-being of human subjects,” as required by the Declaration of Helsinki, researchers should have no reason to expect beforehand that the new treatment will be any better or worse than the old. They should be genuinely uncertain about that (ostensibly the reason for doing the trial in the first place). Otherwise, they would be guilty of deliberately giving an inferior treatment to some of the volunteers—something they must not do if the well-being of their subjects is given precedence. This state of uncertainty (sometimes referred to as “equipoise”) is a generally accepted requirement for the ethical conduct of clinical trials. One of its implications is that researchers may not give placebos to subjects in control groups if there is known to be an effective treatment.
In 1994, research showed that administering an intensive regimen of zidovudine (also called AZT) could prevent pregnant women with HIV from transmitting the infection to their offspring; that regimen quickly became used throughout the US and other developed countries.6 But there was interest in knowing whether a less intensive (and cheaper) regimen of zidovudine would have the same effect, and some reason to believe it might. So the NIH and CDC sponsored a series of clinical trials in developing countries, mainly in sub-Saharan Africa, of a less intensive regimen. But instead of comparing it with the intensive regimen, these trials employed placebos in the control groups. The researchers could have provided the intensive regimen to the control groups, but they believed they would get faster results by using placebos. Yet they knew that by using placebos instead of a known effective treatment, roughly one in six newborns in the control groups would develop HIV infection that could easily have been prevented. (The rate of transmission in untreated women was already fairly well established.) Moreover, the research would have been prohibited in the United States and Europe.
When this became widely known in 1997 as a result of a publication in The New England Journal of Medicine, there was intense controversy, with many people (including me) critical of the trials, and others strongly defending them, including the directors of the NIH and CDC.7 The defenders of the research used versions of two now familiar arguments. First, they said the research stood to save more lives than it cost (the utilitarian argument used in nearly all questionable research) because using a placebo control would yield quicker results, and second, they said that the women in the control groups would not have received effective treatment outside the trials because it wasn’t generally available in that part of the world, so they needn’t get it within the trials (a version of the “condemned anyway” argument).
In the wake of this controversy, some members of the WMA, most importantly the AMA, wanted the fifth revision of the Declaration of Helsinki to approve the use of placebos in clinical trials if the best current treatment was not generally available in the country where the trials were conducted.8 But others argued that researchers are responsible for all the human subjects they enroll, both the experimental and control groups, even if the best clinical care is not generally available in the particular country where the research is done. To the surprise of many, the latter view won the day, and the 2000 revision stated that no one should be given a placebo unless there is no known treatment—regardless of where the research is conducted.
The revision also raised the question of whether sponsors have continuing obligations to the subjects of their research in developing countries, such as providing them with any treatments found effective in trials in which they participated. But the debate did not stop. Instead, two years later, under pressure from the AMA, footnotes were added to the relevant sections that modified them to the extent that the document became internally inconsistent. The NIH and the FDA no longer refer to the Declaration of Helsinki (the FDA instead refers to the International Conference on Harmonisation’s less specific Good Clinical Practice guidelines), but it remains a touchstone in many other countries, despite its inconsistencies, and it is of great historical importance.
—This is the first of two articles.

-
1
The streptomycin results were published in the October 30, 1948, issue of the British Medical Journal, titled “Streptomycin Treatment of Pulmonary Tuberculosis: A Medical Research Council Investigation.” For a later firsthand account of the trial and its implications, see John Crofton, “The MRC Randomized Trial of Streptomycin and Its Legacy: A View from the Clinical Front Line,” Journal of the Royal Society of Medicine, Vol. 99, No. 10 (October 2006). ↩
-
2
Given the thousands of clinical trials conducted every year and the relatively small number of new treatments that become available, it would be impossible for it to be otherwise. In fact, according to FDAReview.org, only 8 percent of all drugs that enter clinical trials are eventually approved for sale by the FDA. ↩
-
3
See The Nazi Doctors and the Nuremberg Code: Human Rights in Human Experimentation, edited by George J. Annas and Michael A. Grodin (Oxford University Press, 1992). This is an excellent history and analysis, which includes the Nuremberg Code and the Declaration of Helsinki up through the 1975 revision. ↩
-
4
Described by Jonathan D. Moreno in Undue Risk: Secret State Experiments on Humans (Routledge, 2000). ↩
-
5
In 1966, Henry Beecher, professor of research anesthesia at Harvard Medical School, published twenty-two examples of unethical human research. Example number sixteen was the Willowbrook study. Beecher’s paper was widely reported, and contributed to the impetus for the 1974 National Research Act. See Henry K. Beecher, “Ethics and Clinical Research,” The New England Journal of Medicine, Vol. 274, No. 24 (June 16, 1966). ↩
-
6
Edward M. Connor, Rhoda S. Sperling, Richard Gelber, et al., “Reduction of Maternal-Infant Transmission of Human Immunodeficiency Virus Type 1 with Zidovudine Treatment,” The New England Journal of Medicine, Vol. 331, No. TK (November 3, 1994). ↩
-
7
See Peter Lurie and Sidney M. Wolfe, “Unethical Trials of Interventions to Reduce Perinatal Transmission of the Human Immunodeficiency Virus in Developing Countries,” The New England Journal of Medicine, Vol. 337, No. 12 (September 18, 1997). For responses, see also Marcia Angell, “The Ethics of Clinical Research in the Third World,” The New England Journal of Medicine, Vol. 337, No. 12 (September 18, 1997); and Harold Varmus and David Satcher, “Ethical Complexities of Conducting Research in Developing Countries,” The New England Journal of Medicine, Vol. 337, No. 14 (October 2, 1997). ↩
-
8
For a discussion of this controversy, see Jonathan Kimmelman, Charles Weijer, and Eric M. Meslin, “Helsinki Discords: FDA, Ethics, and International Drug Trials,” The Lancet, Vol. 373, No. 9,657 (January 3, 2009). ↩

{kind=link}
{kind=link}